
Next generation sequencing (NGS) is now in routine use for a broad range of research and clinical applications. The rapid rate of adoption has been facilitated by falling reagent costs, benchtop instruments, improved chemistries and improved data analysis solutions. However, the cost and complexity of data analysis still remain significant hurdles — particularly for whole genome sequencing. In the majority of cases, targeted approaches, such as custom NGS panels, are more cost-effective and generate significantly less, but equally meaningful data in a much shorter timescale.
Targeted sequencing requires an initial sequence enrichment step, which, if poorly designed, can be a source of bias and error in the downstream sequencing assay1. This application note discusses the main strategies employed to optimise the enrichment step, depending on the type of assay chosen.
Two broad categories of enrichment assays exist: amplicon (PCR) and hybridisation (Figure 1). As a very general rule, hybridisation-based assays, when designed well, offer superior performance2.
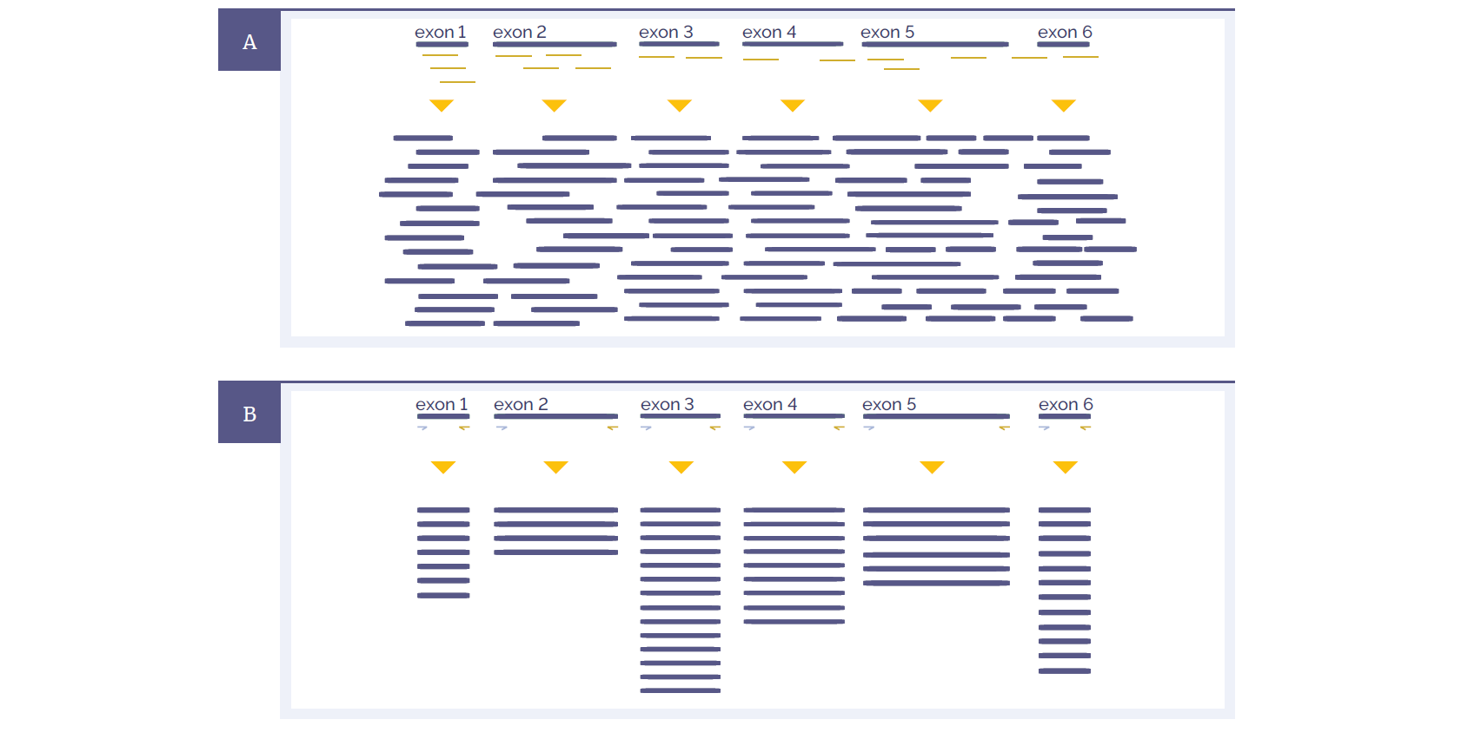
Figure 1: Schematic representations of amplicon and hybridisation enrichment approaches. (A) Hybridisation assays begin with random shearing of the genomic DNA, followed by capture using long oligonucleotide baits. Because of this random shearing, fragments captured are overlapping and unique. Baits can be tiled, overlapped and positioned to overcome challenges of repetitive sequences etc. With advanced design, capture can be made very uniform. (B) Amplicon assays provide less flexibility in the positioning and design of primers – primer pairs need to flank the region to be targeted. All fragments generated from a single primer set are identical, with the disadvantage that assay artefacts cannot be distinguished from genuine variation. Primer competition and preferential amplification of some regions over others will lead to non-uniform enrichment.
Hybridisation protocols start with random shearing of the DNA, followed by “capture” of the randomly sheared overlapping fragments with long oligonucleotide (oligo) baits. This allows independent sequencing of a large number of unique fragments. Any duplicates (assay artefacts) can be easily identified and removed, leaving high-quality data for analysis. Because the fragments are randomly sheared they should not align perfectly with one another and if they do, they are most certainly duplicates. In addition, enrichment of challenging regions such as GC-rich regions or internal tandem repeats can be optimised by careful positioning and design of baits. Long oligo baits can tolerate sequence variation, so that all alleles of a heterogeneous mix can be captured equally. Amplicon assays require design of primers flanking the region to be amplified. The resulting amplification products are identical, such that duplicates cannot be distinguished from unique products.
In making the choice between hybridisation and amplicon approaches, there are several factors which are worth considering (Figure 2).

Figure 2: A number of key factors are important in selection of the most appropriate enrichment assay for a given application.

Figure 3: Hybridisation-based enrichment delivers more uniform coverage of GC-rich regions. Comparison of amplicon and hybridisation-based enrichment of the GC-rich exons 4 and 5 of the TP53 gene.

Figure 4: A deletion within the amplicon can cause preferential amplification of smaller fragments.

Figure 5: Single nucleotide variants (SNVs) in primer sites can lead to allelic bias and drop-out.
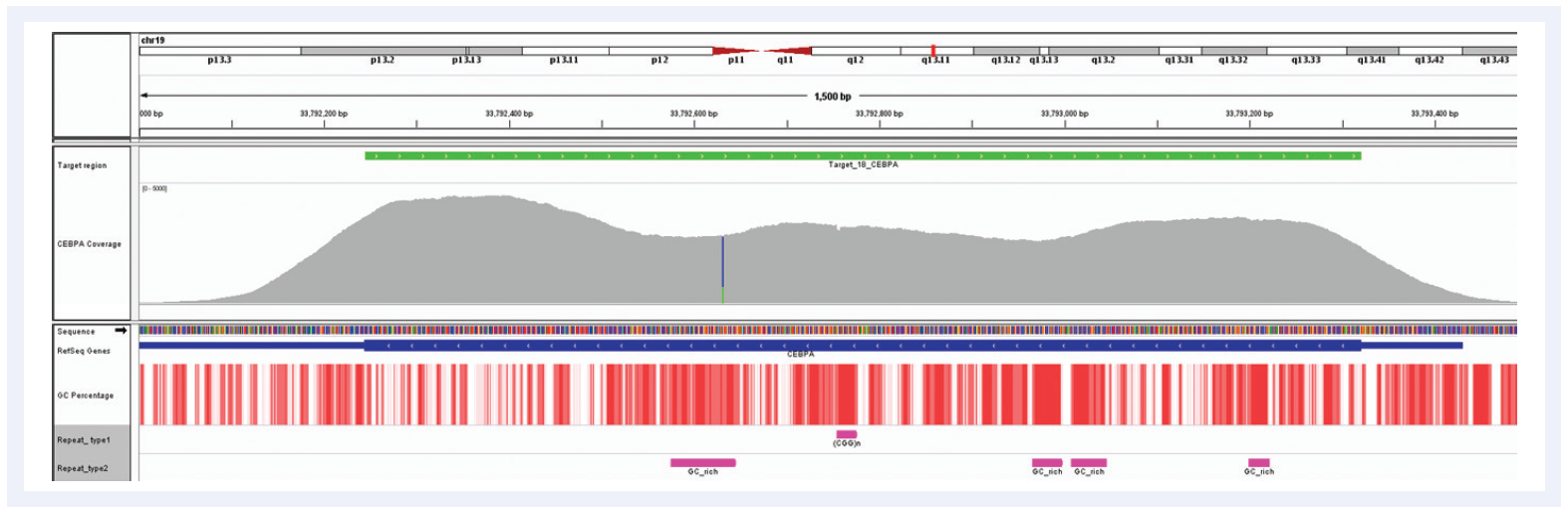
Figure 6: Despite the very high GC content of the CEBPA gene and the number of repeat regions, hybridisation enrichment coupled with expert bait design can still achieve excellent results. Depth of coverage per base (grey). Targeted region (green). Gene coding region as defined by RefSeq (blue). GC percentage (red). Repeat regions, and those rich in GC (pink). Data generated using SureSeq myPanel™ custom content.
Samples vary in quality and quantity so any assay must be able to deal with a wide range of input DNA types and input quantities:

Figure 7: The SureSeq™ FFPE DNA Repair Mix significantly improves mean target coverage resulting in more confident calls. Data obtained using 500 ng of FFPE DNA from ovarian and colon cancer samples; 16 samples per MiSeq® lane.

Figure 8: Effect of reduced amount of DNA input on mean target coverage (A) and %OTR (B).
Performance should be a key requirement for all applications: the confidence that the assay will detect all variants present in any region of interest, while avoiding false negatives and false positives. Hybridisation assays offer a number of key benefits which enhance performance:
The ultimate goal of any sequencing assay is to discover all variants present. Uniformity of enrichment means that all regions are represented more equally, and that variants present in any region will be called (Figure 9). It also allows much lower average sequencing depths to be used, enabling larger numbers of samples to be multiplexed in a run, and significant cost savings.

Figure 9: High uniformity of coverage allows the reliable detection of low frequency somatic indels even in FFPE derived DNA. Example is of 12 bp deletion (c.754_765delCTCACCATCATC) in TP53, 6% deletion, mean target coverage >1400, 12 samples per MiSeq lane, using the SureSeq Ovarian Cancer Panel.
Uniformity is particularly important when looking at heterogeneous samples. For example tumour mixed with normal tissue, or somatic variants present only within a single clone in a heterogeneous tumour sample where it is essential to have enough reads to confidently call a variant at any given position.
As NGS moves forwards, the aspiration is clear: confident calling of all variants present with no false negatives and no false positives. The most common reason for missing variants (i.e. false negatives) is lack of coverage at the variant locus due to non-uniform enrichment, the importance of which is illustrated with the example of exon 9 of CALR gene, an important exon for interrogation of myeloproliferative disorders (Figures 10 and 11).
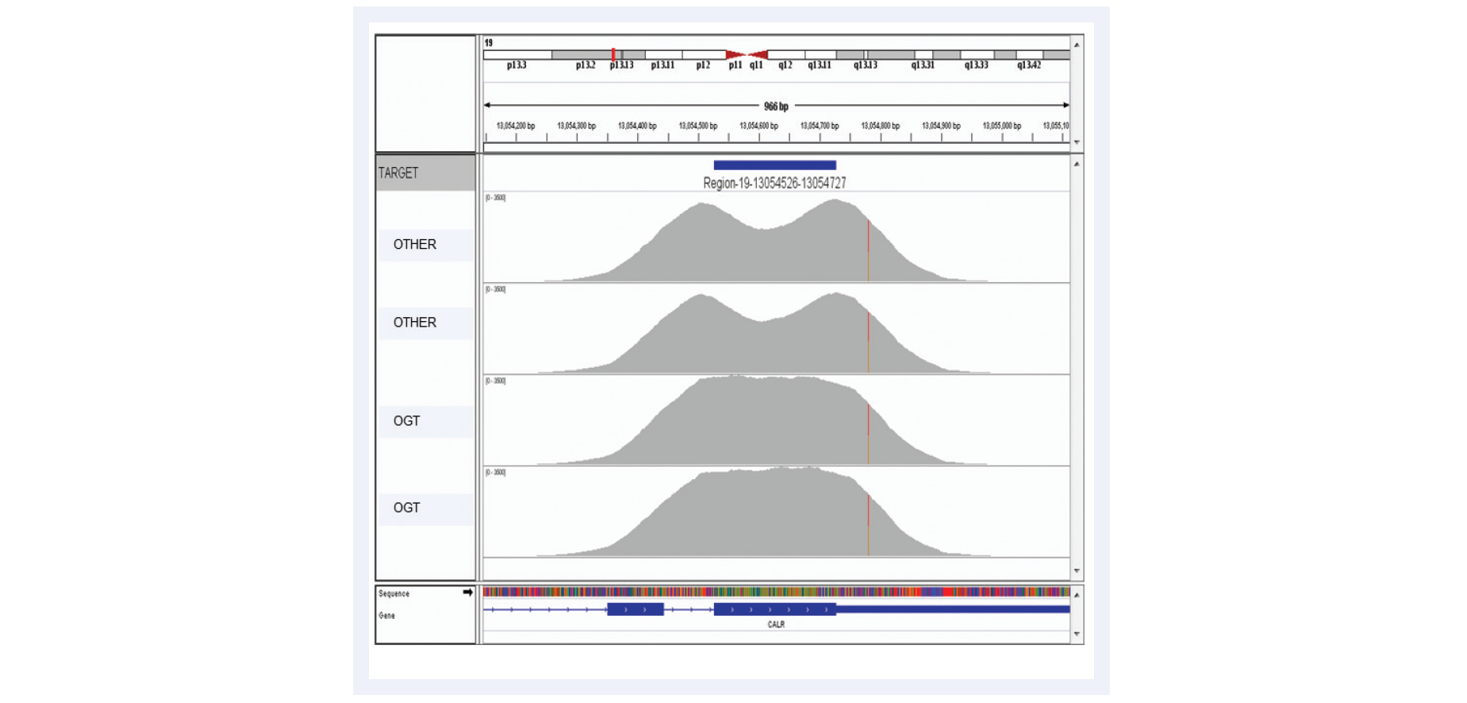
Figure 10: Advanced bait design strategies deliver uniform enrichment, reducing the likelihood of false negative results. The top two captures have been completed using baits designed with standard commercially available software. They have a considerable dip in coverage in the middle of the exon due to the fact it presents a low complexity region with low nucleotide diversity. Most algorithms would avoid such regions in the design. However, OGT’s superior bait design can increase the evenness of coverage of such regions.
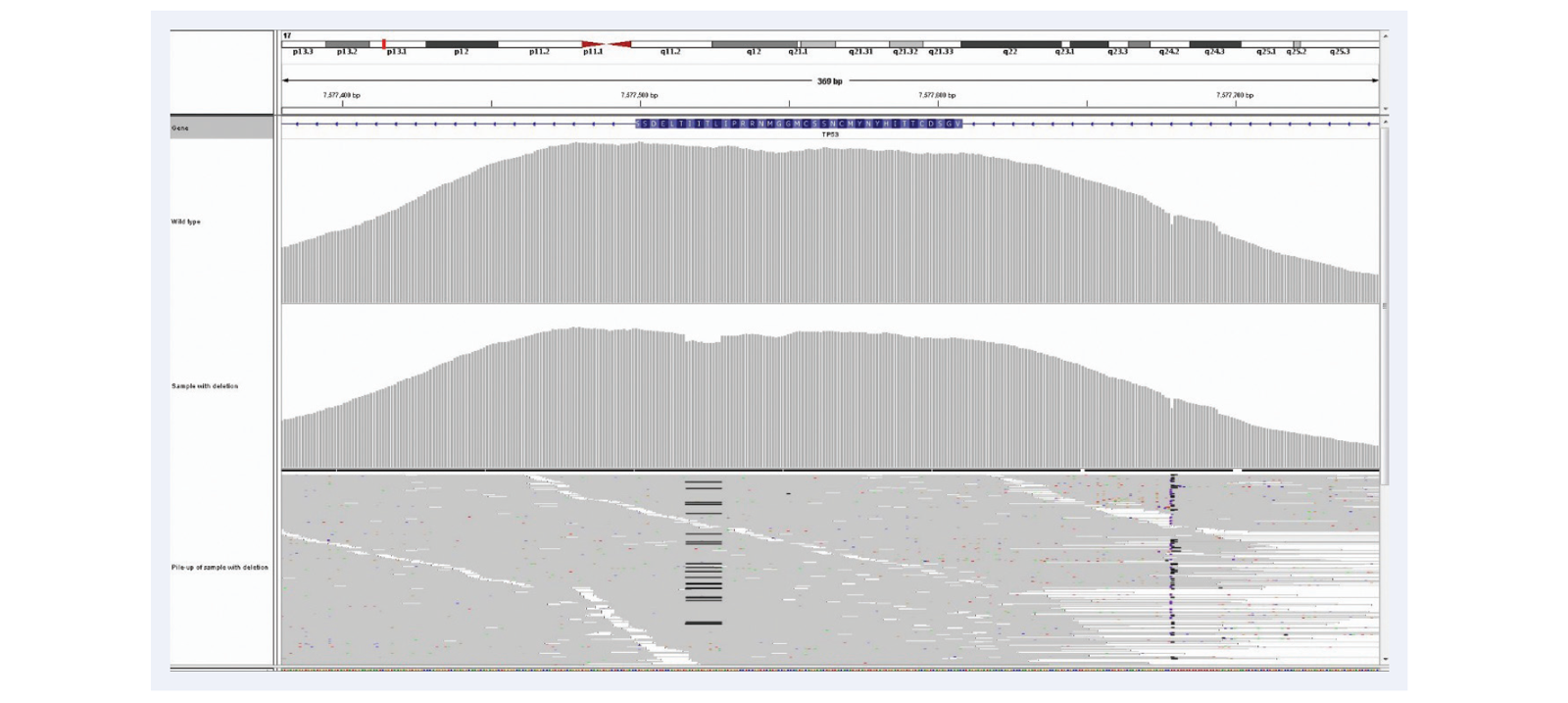
Figure 11: Superior uniformity of coverage allows reliable detection of indels even as big as 1/3 of a read length. Shown here 52bp deletion in exactly the same region as illustrated in Figure 10, exon 9 of the CALR gene. 23% deletion (c.1092_1143del_52bp). Mean target coverage >1000, 24 samples per MiSeq lane, using the SureSeq Myeloid panel.
The highlighted box below provides a summary of all of the potential sources of error and bias. As a very broad rule, hybridisation-based assays offer greater opportunity for optimisation through probe design and placement, and they can offer better uniformity of coverage, fewer false positives, and superior variant detection due to fewer PCR cycles. Hybridisation-based assays also offer greater scope in terms of the number of genes and regions that can be targeted.
The choice of enrichment assay for targeted sequencing assays is an important consideration. No enrichment technology is perfect for every application. The choice will depend largely on the size of region to be targeted, cost of sequencing and the required coverage uniformity and sensitivity of the assay. Optimisation is also important and hybridisation-based assays offer more scope for superior performance through optimisation of bait design.
Even proof-reading PCR polymerases introduce errors. The likelihood of artefacts (as well as the rate of duplication) increases with increasing PCR cycles. Amplicon-based assays are reliant entirely on PCR, and are potentially more susceptible to artefacts than hybridisation-based assays, which aim to minimise the number of PCR cycles.
Duplicates are amplification artefacts, normally arising during the library preparation stage. It is highly desirable to remove them prior to data analysis, otherwise some regions may be massively over-represented. In a hybridisation-based assay, duplicate reads can be identified easily and removed. However, in a PCR assay, not only is it impossible to remove duplicates during the analysis step, but it is also true that there are generally more duplicates due to the enrichment step. Duplicates are a particular problem when input quantities of DNA are limiting, because higher numbers of PCR cycles need to be used.
PCR is difficult to multiplex where consistent and uniform amplification of each region is desired. There are thought to be two main processes that introduce amplification bias during PCR, PCR selection and PCR drift4. In PCR selection, some amplicons are favoured and therefore over-represented due to intrinsic properties of the target sequence, flanking sequences or genome composition. Key contributors to this type of variation include preferential denaturation and amplification of low GC content templates, higher binding efficiency of GC-rich primers (particularly when using degenerate primers) and direct correlation between amplification efficiency and gene copy numbers. PCR drift is assumed to be caused by random interaction of the components of the mix early on in the amplification when the original genomic material is still the main source of template. This type of variation is variable between reactions and more difficult to control. There are ways to optimise PCR that will reduce bias1 including increasing the amount of template, reducing the number of cycles, optimising the instrumentation and performing the multiplex in a number of discrete, lower-plex reactions which are then pooled after amplification.
Variant positional bias is a particularly important consideration for amplicon assays where there is some constraint in choice of position for primer sites. If a SNP or variant happens to fall within the primer site itself, it will not be detected. This is less likely to be an issue with hybridisation-based methods that use long oligonucleotides that can tolerate target sequence variation and can be tiled across the region to be enriched.
Repeat regions and pseudogenes represent significant challenges for all enrichment technologies. Depending on the size of the region, hybridisation-based assays may allow better targeting of these regions, by allowing design of baits to flanking regions.
SureSeq™: For Research Use Only; Not for Diagnostic Procedures. This document and its contents are © Oxford Gene Technology IP Limited – 2021. All rights reserved. Trademarks: OGT™, SureSeq™ and myPanel™ (Oxford Gene Technology); Agilent® (Agilent Technologies Inc.); MiSeq® (Illumina Inc.).
Call +44 (0)1865 856800 Email contact@ogt.com
Send us a message and we will get back to you
 Visit USA site
Visit USA site Visit Canada site
Visit Canada site